Idiopathic Pulmonary Fibrosis
On this page
Overview
Idiopathic pulmonary fibrosis, or IPF, is a condition that causes progressive scarring of the lungs. Fibrous scar tissue builds up in the lungs over time, affecting their ability to provide the body with enough oxygen. The cause of the condition is unknown.
IPF affects more than 100,000 people in the United States, with 30,000 to 40,000 new cases diagnosed each year. Typically the disease is found in people between the ages of 50 and 70 and affects men more frequently than women. Most patients are former smokers. There are no proven risk factors for IPF, but a minority of patients have a family history of lung scarring.
Our approach to idiopathic pulmonary fibrosis
UCSF offers specialized care for all types of interstitial lung disease, including idiopathic pulmonary fibrosis. Treatments include medication to slow the decline in lung function, care for other health problems that often affect patients with IPF, and a special exercise and education program designed for patients with chronic lung disease. Our specialists review each case as a team to ensure that every patient gets the right diagnosis and most effective care.
For patients who get worse despite treatment, lung transplantation may be an option. UCSF is home to a high-performing lung transplant program with the expertise to handle the most complex, challenging cases.
Signs & symptoms
Symptoms of IPF often appear gradually and include:
- Shortness of breath, particularly during or after physical activity
- Chronic, dry hacking cough
- Crackle sound in the lungs heard through a stethoscope
- Rounding of the fingernails, a condition called clubbing
Symptoms of IPF may mimic those of other diseases that cause lung scarring, so diagnosing IPF often involves ruling out other conditions. Several visits with your doctor may be needed to finalize your diagnosis and treatment approach.
Diagnosis
Diagnosing idiopathic pulmonary fibrosis requires input from pulmonologists, radiologists and, in many cases, pathologists experienced in evaluating patients with interstitial lung disease. A face-to-face discussion among these various specialists is often necessary to make an accurate diagnosis.
To determine if you have IPF, your doctor will start by conducting a thorough medical history and physical examination. The medical history will include discussing other medical problems you have that could be related to lung scarring, such as connective tissue diseases, and reviewing any medications you're taking. The physical exam will include listening to your chest with a stethoscope to check for a crackling sound and carefully examining your skin and joints.
In addition to a thorough medical history and physical exam, your doctor may conduct the following tests:
- Pulmonary function test (PFT). This test involves a series of breathing exercises that measure the airflows, volume of air in your lungs and ability of your lungs to extract oxygen from the air. This allows your doctor to assess the function of your lungs.
- High resolution computed tomography (HRCT). This is a special type of CT scan that provides your doctor with high-resolution images of your lungs. Images are taken in several different ways, including lying on your back (supine), lying on your chest (prone) and while breathing air out of your chest (dynamic expiration). These images are extremely valuable in determining whether or not you have IPF. Having an HRCT is very similar to having a regular CT scan; they both take only a few minutes.
- Blood tests. We may order blood tests, also called serologies, to look for evidence of connective tissue diseases such as rheumatoid arthritis or scleroderma. Some patients with these connective tissue diseases develop lung problems before the more typical signs and symptoms appear.
- Six-minute walk test. This test evaluates the distance you can walk within six minutes and the oxygen saturations, measured by finger or ear probe, you achieve while walking.
- Bronchoscopy. In this test, the doctor passes a flexible fiberoptic scope, about the diameter of a pencil, into the lungs to take fluid and tissue samples. This test doesn't require an overnight stay in the hospital. It's unclear if bronchoscopy provides any benefit in diagnosing IPF, but it may be performed in certain circumstances.
- Surgical lung biopsy. Some patients need this test to definitively diagnose IPF. A cardiothoracic surgeon performs the surgical lung biopsy, usually with small tools and cameras through one-inch-long incisions. Patients will need to stay in the hospital for a few days.
Treatments
Two antifibrotic medications – nintedanib and pirfenidone – were approved in the fall of 2014 for use in idiopathic pulmonary fibrosis. While these medications are not a cure, they have both been shown to slow the decline of lung function over time.
Ongoing studies of other medications for IPF have shown initial promise, but need more research.
Pulmonary rehabilitation is a structured exercise and education program designed to increase endurance, strength, and exercise capacity, as well as to decrease fatigue and shortness of breath, for people with chronic lung disease, including people with IPF.
Additionally, it is important that any other medical problems that are associated with IPF are treated, such as gastroesophageal reflux disease (GERD) and pulmonary hypertension.
Finally, lung transplant may be an effective treatment option for some people with IPF.
Awards & recognition

Best in the West and No. 3 in the nation for pulmonology & lung surgery

Designated pulmonary fibrosis care center
Recommended reading

FAQ: Cyclophosphamide
Cyclophosphamide is a medication that suppresses the immune response and reduces lung inflammation. Find answers to common cyclophosphamide questions.
FAQ: Methotrexate
Find answers to common questions about methotrexate, an immunosuppressant, including what to know before taking the drug and potential side effects.
FAQ: Mycophenolate
Get answers to questions about mycophenolate, an immunosuppressant, including what to know before taking the drug, side effects and necessary monitoring.
FAQ: Prednisone
Get information about prednisone, a medication to treat lung inflammation, including precautions, side effects and necessary monitoring.
GERD in ILD Patients
Interstitial lung disease (ILD) may be linked to gastroesophageal reflux disease (GERD). Understand the symptoms of GERD, how it’s diagnosed and treatments.
ILD Nutrition Manual
Check out our nutrition guide for people with interstitial lung disease (ILD). Eating well can help prevent infection and keep your respiratory system healthy.
ILD Patient Resources
Stay informed with these interstitial lung disease (ILD) resources, including support groups, medication information, associated conditions and more.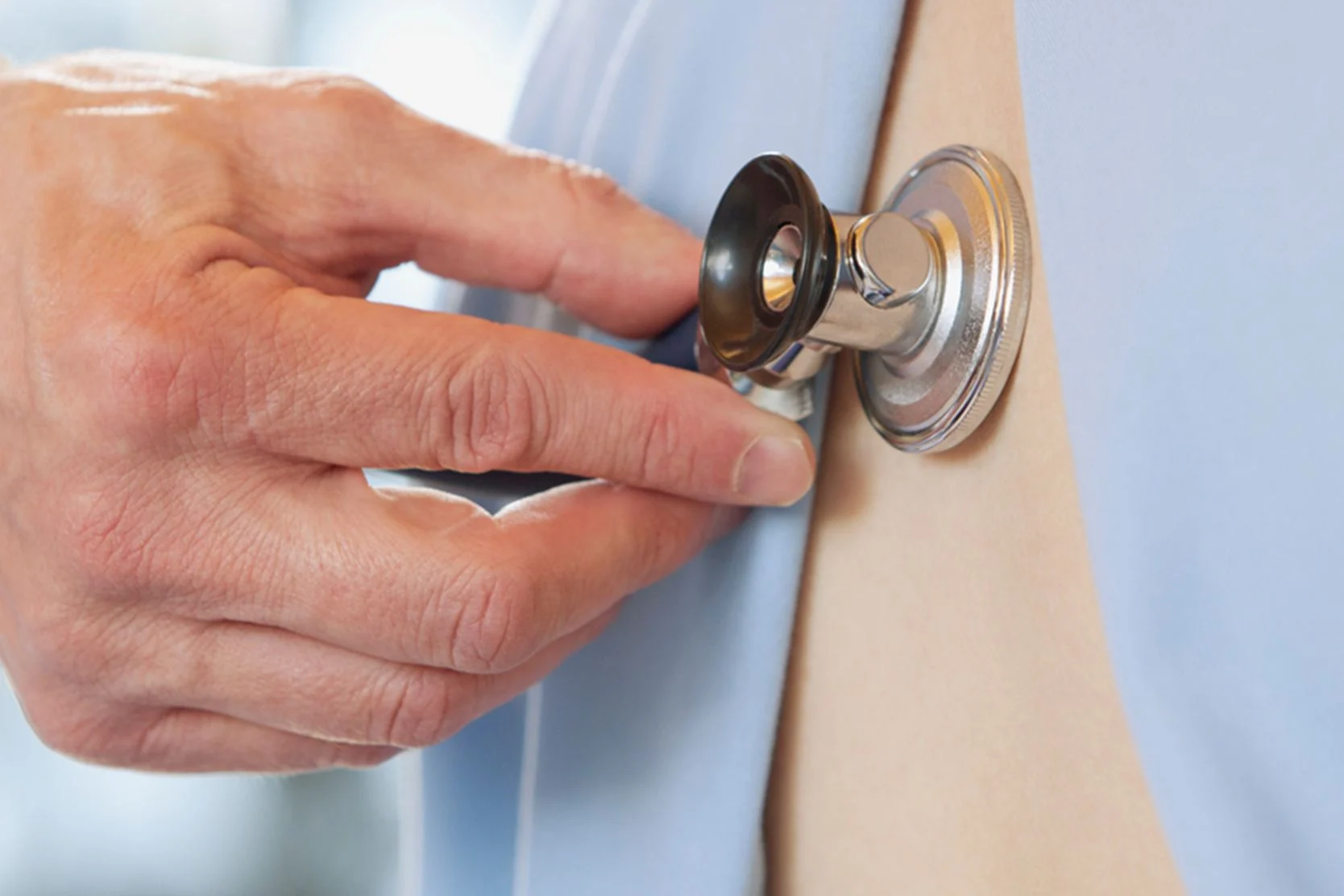
Pulmonary Hypertension and Interstitial Lung Disease
Pulmonary hypertension (PH) is common in patients with interstitial lung disease (ILD). Get information on PH, including diagnosis and treatments.
Pulmonary Rehabilitation for ILD
Our respiratory therapists provide pulmonary (lung) rehabilitation for people with ILD in the Bay Area. We help reduce symptoms and improve quality of life.
Supplemental Oxygen
Explore our comprehensive guide to using supplemental oxygen, including safety information and travel recommendations.
Related services & treatments
Specialties
Treatments
- Pulmonary and Cardiac Rehabilitation
- Lung Transplant
Helpful resources
Support services

Case Management and Social Work
Connect with a team that can help you find resources, solve problems and advocate for you during treatment at UCSF.
Mindfulness-Based Stress Reduction Class
This eight-week class teaches mindfulness practices that can reduce stress and improve your overall health, such as meditation and body awareness.
Patient Relations
We welcome feedback about your experience at UCSF Health. Find out how to contact us with comments, questions or concerns.
Pulmonary Support Groups
Find support groups for people with pulmonary conditions such as obstructive sleep apnea, interstitial lung disease (ILD) and pulmonary hypertension.
Spiritual Care at UCSF
Chaplains representing many faiths are available around the clock to provide support, comfort and counsel to patients, families and caregivers.
UCSF Health medical specialists have reviewed this information. It is for educational purposes only and is not intended to replace the advice of your doctor or other health care provider. We encourage you to discuss any questions or concerns you may have with your provider.